Usecase ea: Difference between revisions
No edit summary |
No edit summary |
||
| Line 1: | Line 1: | ||
=== | {{DISPLAYTITLE: }} | ||
One of the standard approaches in downstream analysis of gene expression is enrichment analysis that can be useful for evaluating biological function under condition. In this use-case, two | === <center> Annotation Enrichment Analyses </center> === | ||
---- | |||
One of the standard approaches in downstream analysis of gene expression is enrichment analysis that can be useful for evaluating biological function under condition. In this use-case, two scenarios of annotation enrichment analyses are introduced for refTSS users. | |||
* ''' Standard enrichment analyses''' | |||
: '''GO and KEGG pathway annotation enrichment analysis''' are commonly used for examination of the over-representation of gene annotations. Here we introduce the workflow of general annotation enrichment analysis using refTSSs. | |||
GWAS-LD enrichment analysis | * ''' GWAS-LD enrichment analysis''' | ||
: Novel enrichment service called '''refTSS-LD''' was lunched on refTSS4. refTSS-LD provides '''GWAS-LD enrichment analysis''' using refTSS IDs. Here is an example input file of TSS ID lists from CAGE analysis of neural crest cell differentiation experiments with iPS cells ([[Source::https://reftss.riken.jp/reftss/userfiles/gwas_ld_example_ids.230607.txt| day0_vs_day18]]). | |||
---- | |||
<html> | <html> | ||
| Line 10: | Line 18: | ||
</html> | </html> | ||
==== 1. General annotation enrichment analysis ==== | |||
Annotation enrichment analysis examines the over-representation of genes of interest with biological annotations such as GO and KEGG pathways. To apply TSS analysis using refTSS to general annotation enrichment, TSS IDs must be converted to gene IDs or symbols for input into annotation enrichment tools. refTSS4 has newly launched a simple web tool for converting TSS IDs to gene symbols to support refTSS users. | |||
<html> | |||
<p style="text-align: center;"><img src="https://github.com/suimye/test_github/assets/296176/bca5220d-ebd4-48a2-a923-079112ce2a64" style="zoom:60%;"></p> | |||
</html> | |||
===== 2. GWAS-LD enrichment analysis ===== | # TSS tag counts are calculated against the refTSSv4 TSS reference using TSS sequencing data (e.g., CAGE, RAMPAGE, etc.). The `featureCounts` function in the Subread package can be useful for this process. | ||
# Normalization and statistical testing were performed by standard packages such as DESeq2 and edgeR. For example, edgeR is used for statistical significance calculations between treated and control samples. | |||
# The IDs of TSSs showing significant expression changes are extracted from the differential expression analysis results. | |||
# The TSS IDs are input into the ID conversion tool on the refTSS website. | |||
<html> | |||
<p style="text-align: center;"><img src="https://github.com/suimye/test_github/assets/296176/7d0d22ab-4622-4d9f-819d-0c0df08e42db" style="zoom:60%;"></p> | |||
</html> | |||
# The extracted Gene Symbols are input into external enrichment analysis tools such as '''DAVID, MetaScape, and ClusterProfiler'''. | |||
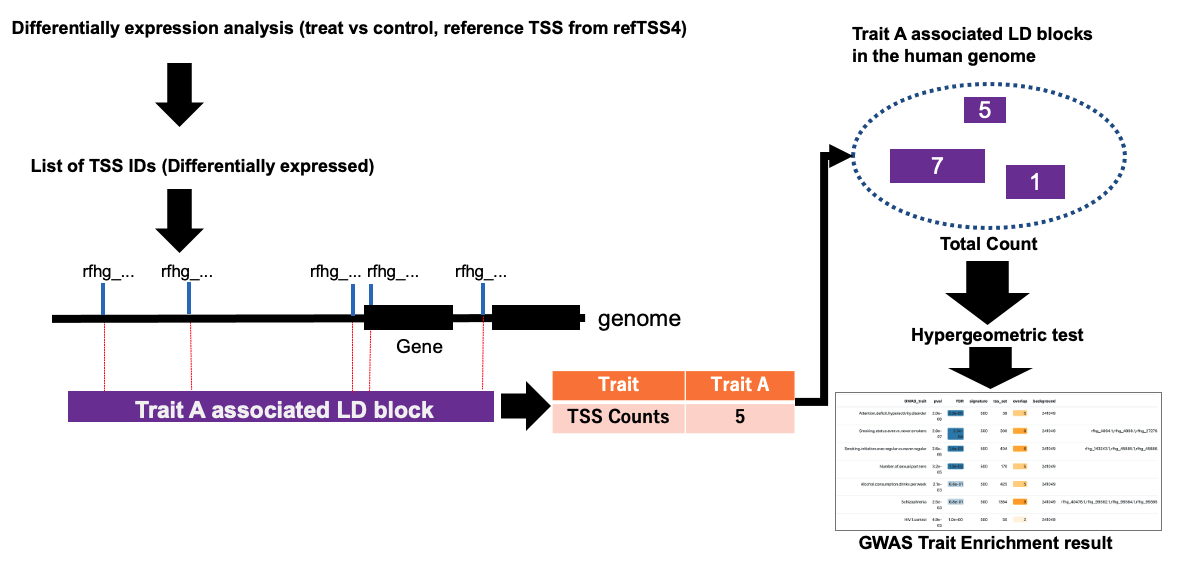
===== 2. GWAS-LD enrichment analysis on refTSS-LD ===== | |||
<html> | <html> | ||
| Line 33: | Line 53: | ||
---- | ---- | ||
Revision as of 15:14, 7 October 2024
Annotation Enrichment Analyses
One of the standard approaches in downstream analysis of gene expression is enrichment analysis that can be useful for evaluating biological function under condition. In this use-case, two scenarios of annotation enrichment analyses are introduced for refTSS users.
- Standard enrichment analyses
- GO and KEGG pathway annotation enrichment analysis are commonly used for examination of the over-representation of gene annotations. Here we introduce the workflow of general annotation enrichment analysis using refTSSs.
- GWAS-LD enrichment analysis
- Novel enrichment service called refTSS-LD was lunched on refTSS4. refTSS-LD provides GWAS-LD enrichment analysis using refTSS IDs. Here is an example input file of TSS ID lists from CAGE analysis of neural crest cell differentiation experiments with iPS cells (day0_vs_day18).
1. General annotation enrichment analysis
Annotation enrichment analysis examines the over-representation of genes of interest with biological annotations such as GO and KEGG pathways. To apply TSS analysis using refTSS to general annotation enrichment, TSS IDs must be converted to gene IDs or symbols for input into annotation enrichment tools. refTSS4 has newly launched a simple web tool for converting TSS IDs to gene symbols to support refTSS users.
- TSS tag counts are calculated against the refTSSv4 TSS reference using TSS sequencing data (e.g., CAGE, RAMPAGE, etc.). The `featureCounts` function in the Subread package can be useful for this process.
- Normalization and statistical testing were performed by standard packages such as DESeq2 and edgeR. For example, edgeR is used for statistical significance calculations between treated and control samples.
- The IDs of TSSs showing significant expression changes are extracted from the differential expression analysis results.
- The TSS IDs are input into the ID conversion tool on the refTSS website.
- The extracted Gene Symbols are input into external enrichment analysis tools such as DAVID, MetaScape, and ClusterProfiler.
2. GWAS-LD enrichment analysis on refTSS-LD
From the menu, select refTSS-LD and paste the TSS IDs into the box.
3. Interpreting the results of the enrichment analysis
The table shows the results of the GWAS-LD analysis conducted using the example data. Results are sorted by order of FDR. The enriched TSS IDs are displayed in the "hits" column, allowing for inspection of which TSSs are concentrated in LD blocks grouped by traits.